Diabetic Retinopathy and Nephropathy May Not Progress Together in Type 2 Diabetes, Reveals Study
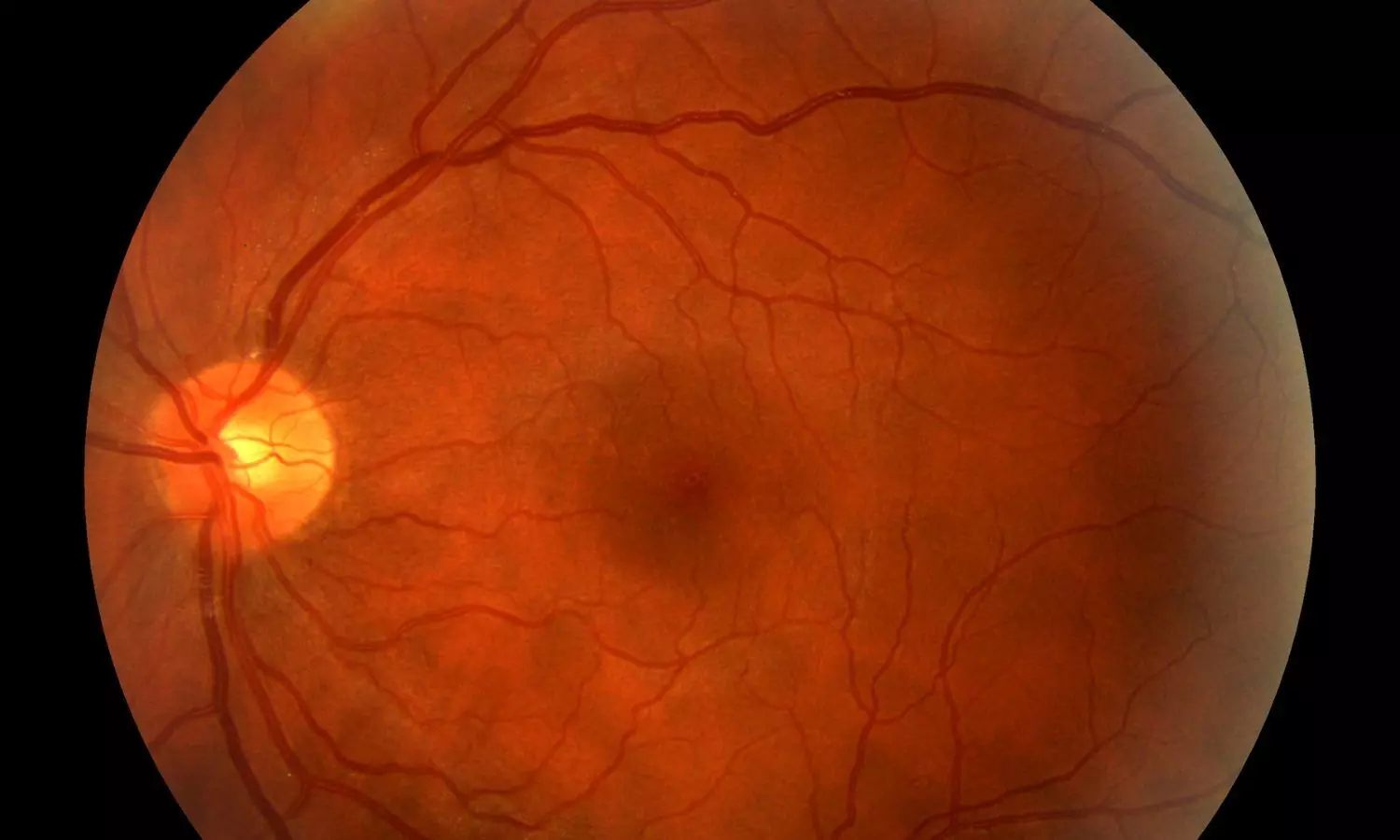
USA: A recent study published in Frontiers in Endocrinology has shed light on a critical finding: diabetic retinopathy (DR) and diabetic nephropathy (DN)—two major microvascular complications in type 2 diabetes—do not always progress in parallel. Conducted by Dr. Ashley Fitzgerald and colleagues from the Department of Ophthalmology and Visual Sciences, University of New Mexico School of Medicine, the cross-sectional case-control study aimed to evaluate the degree of concordance between the severity of DR and DN in a New Mexican diabetic population.
To explore this relationship, researchers analyzed data from two distinct patient groups—one with end-stage renal disease (ESRD, Stage 5, on dialysis) and the other with early-stage DN (Stage 1 or 2). The ESRD group included 164 individuals with severely reduced kidney function (eGFR <15 mL/min), while the mild DN group comprised 165 patients with relatively preserved kidney function (eGFR >60 mL/min). Retinal status was evaluated through dilated fundus examinations, and systemic clinical parameters were extracted from retrospective chart reviews.
The following were the key findings of the study:
- 65% of patients with end-stage renal disease (ESRD) exhibited proliferative diabetic retinopathy (PDR), the most advanced stage of DR.
- 18% of patients in the ESRD group had either mild or no signs of diabetic retinopathy.
- In the mild diabetic nephropathy (DN) group, 38% of individuals already showed signs of PDR.
- These findings highlight a lack of consistent overlap between the progression of diabetic kidney disease and eye complications.
- Higher systolic and diastolic blood pressure were significantly associated with the presence of PDR, particularly among ESRD patients.
- Elevated LDL cholesterol levels were linked to an increased risk of PDR.
- Increased levels of albuminuria and serum creatinine were also significantly associated with PDR.
- HbA1c levels, a marker of glycemic control, showed no significant correlation with PDR severity.
- Younger age was a contributing factor to the development of PDR.
Despite its findings, the study acknowledges several limitations, including its retrospective nature and missing data from patient records, which may introduce selection bias. Some patients had irregular follow-ups, making it difficult to capture the full progression of complications. Additionally, race and ethnicity were self-reported and inconsistently recorded, potentially affecting statistical interpretations.
The researchers concluded that while DR and DN share several modifiable risk factors, their progression is not necessarily synchronous. Relying solely on glycemic control is inadequate for predicting the severity of either condition. Therefore, both complications should be assessed independently, taking into account individual clinical profiles and risk markers. The team also hypothesized that genetic variability may play a role in the discordant progression patterns, with some individuals being genetically predisposed to more aggressive forms of one complication over the other.
“The study emphasizes the importance of individualized assessment and the need for further longitudinal research to better understand the mechanisms underlying the divergence between diabetic eye and kidney disease progression,” the researchers wrote.
Reference:
Fitzgerald, A., Das, R., Moezzi, C. J., Salazar, S. R., Mankad, R., Qualls, C. R., Cabrera, A., Kathuria, A., Monickaraj, F., Tzamaloukas, A., & Das, A. (2025). Discordance of diabetic retinopathy severity in a cohort of diabetic nephropathy patients: A cross-sectional case-control study in a new Mexican population of type 2 diabetes. Frontiers in Endocrinology, 16, 1638415. https://doi.org/10.3389/fendo.2025.1638415
Powered by WPeMatico